Abstract
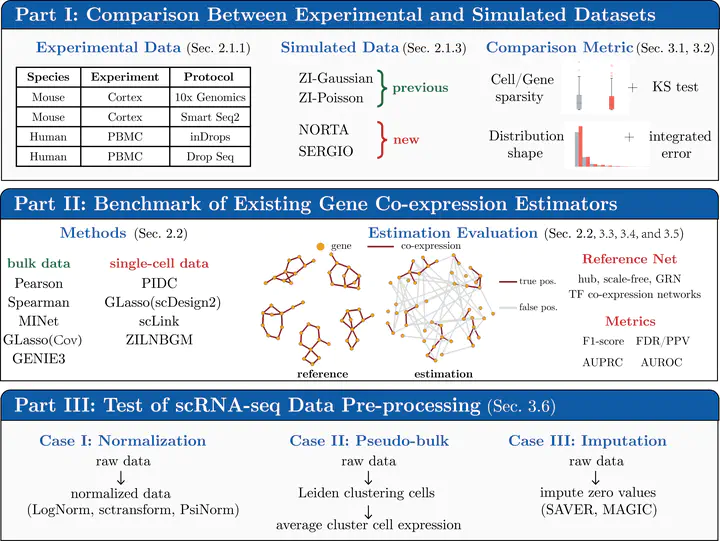
With the rapid advance of single-cell RNA sequencing (scRNA-seq) technology, understanding biological processes at a more refined single-cell level is becoming possible. Gene co-expression estimation is an essential step in this direction. It can annotate functionalities of unknown genes or construct the basis of gene regulatory network inference. This study thoroughly tests the existing gene co-expression estimation methods on simulation datasets with known ground truth co-expression networks. We generate these novel datasets using two simulation processes that use the parameters learned from the experimental data. We demonstrate that these simulations better capture the underlying properties of the real-world single-cell datasets than previously tested simulations for the task. Our performance results on tens of simulated and eight experimental datasets show that all methods produce estimations with a high false discovery rate potentially caused by high-sparsity levels in the data. Finally, we find that commonly used pre-processing approaches, such as normalization and imputation, do not improve the co-expression estimation. Overall, our benchmark setup contributes to the co-expression estimator development, and our study provides valuable insights for the community of single-cell data analyses. Codes are available at https://github.com/rsinghlab/scRNAseq_Coexpression_Benchmark.
Jiaqi Zhang
PhD Student at Computer Science
My research interests include machine learning and bioinformatics.